Research Content
Functional Analysis of ALS2, the Causative Gene for ALS Type 2
What is the ALS2 Gene?
The ALS2 gene, located in the q33 region of the long arm of chromosome 2, consists of 34 exons with a total length of approximately 80kb. It is the causative gene for three types of autosomal recessive motor neuron diseases (ALS2, PLSJ, and IAHSP). The ALS2 gene produces two transcripts of 6.5kb and 2.6kb, and it is strongly expressed in the nervous system, muscles, and kidney tissues. The transcription sequence of ALS2 includes a coding region for a protein of approximately 1657 amino acid residues (184 kDa).
What is the ALS2 Protein?
The ALS2 protein, also known as "Alsin," contains three independent guanine nucleotide exchange factor (GEF)-like domains (RLD, DH/PH, and VPS9). The RLD (regulator of chromosome condensation 1–like domain) at the N-terminus has a β-propeller structure with seven blades, and blade 5 is interrupted by the insertion of an intrinsically disordered region (IDR). The DH/PH (Dbl-homology and pleckstrin-homology) domain downstream of RLD does not have RhoGEF activity but contains eight MORN (membrane occupation and recognition nexus) motifs involved in membrane binding. The C-terminus contains the VPS9 (vacuolar protein sorting 9) domain, which catalyzes the activation of Rab5.

Function of ALS2
Previous research has revealed several aspects of the structure and function of the ALS2 protein:
- ALS2 is an activator of the small G protein Rab5.
- ALS2 plays a crucial role in regulating endosome dynamics through the activation of Rab5 in cultured cell lines, including neurons.
- ALS2 is a regulatory factor involved in axonal elongation of neurons.
- Through Rac1 activation, ALS2 localizes to endosomes via macropinocytosis.
- ALS2 regulates protein degradation through autophagy.
- ALS2 forms homotetramers, and its normal higher-order structure is essential for endosomal localization and Rab5GEF activity.
- Loss of ALS2 function exacerbates the disease symptoms in a mutant SOD1-expressing ALS mouse model.
- ALS2 binds to the small G protein Rab17, controlling the maturation and recycling of Rab17-positive endosomes.
- The intrinsically disordered region (IDR) inserted in the N-terminal RLD of ALS2 controls its intracellular localization and complex formation.
- ALS2 forms a specific high-molecular-weight complex in the central nervous system.
Future Challenges for ALS2
Currently, numerous factors binding to ALS2 have been identified. The future goal is to elucidate the molecular mechanisms of ALS2 structure and activation, as well as the functional connections with novel binding partners and the regulation of endosome-lysosome system function. This will contribute to understanding the mechanism of motor neuron diseases caused by ALS2 dysfunction.
Functional Analysis of ALS-Related Factor SQSTM1/p62
What is SQSTM1/p62?
SQSTM1 (sequestosome-1 or p62) functions as an adapter protein in the autophagy pathway, involved in the degradation pathway of proteins and organelles. It also promotes oxidative stress response through interaction with Keap1 and Nrf2. Additionally, SQSTM1 is known to be involved in the induction pathway of apoptosis through the aggregation of ubiquitinated caspase-8 (refer to the figure below). SQSTM1 accumulates along with ubiquitinated proteins in the spinal cords of the majority of sporadic ALS patients.
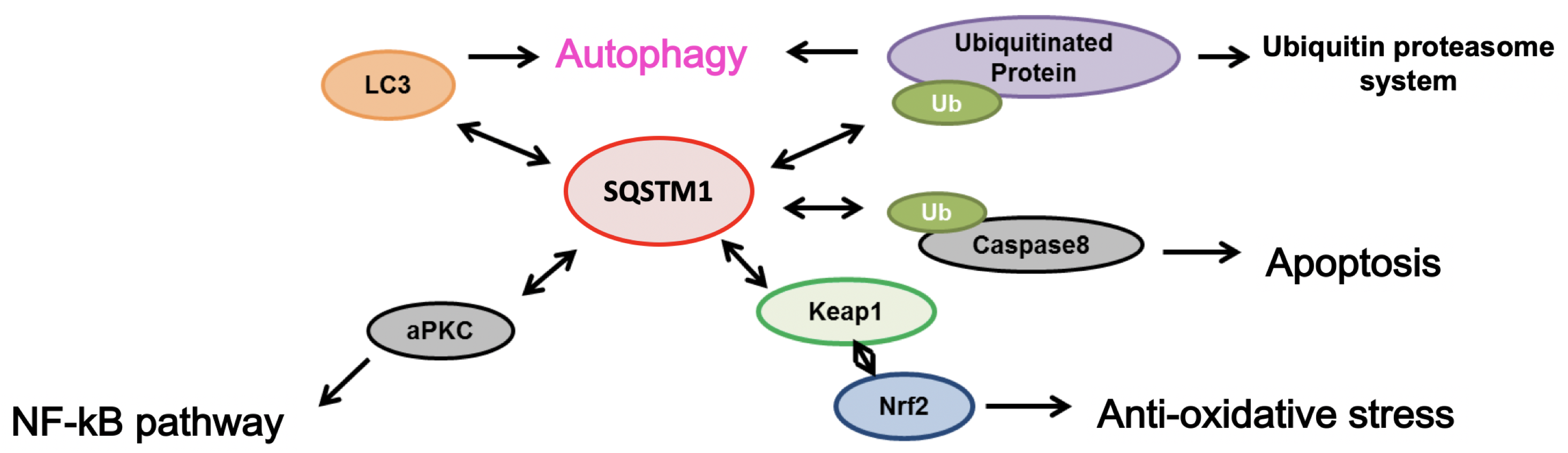
SQSTM1 Gene Mutations and ALS
In recent years, numerous SQSTM1 gene mutations have been identified not only in familial ALS but also in sporadic ALS and frontotemporal dementia (FTD) patients. Through collaborative research with Sichuan University and Peking University in China, we have identified several new SQSTM1 mutations in sporadic ALS patients.
Role of SQSTM1/p62 in Mutant SOD1-Expressing ALS Mouse Model
Past studies have shown that the loss of SQSTM1 function significantly worsens the disease symptoms of the mutant SOD1-expressing ALS mouse model (SOD1H46R). Systemin overexpression of SQSTM1 accelerates the weight loss of SOD1H46R and promotes early onset of the disease. SQSTM1, while promoting aggregate formation in neurons, exhibits neuroprotective effects but can induce neurotoxicity when excessively present. The early onset of the disease is shown to be accelerated additively by the deficiency of both ALS2 and SQSTM1.
Functional Analysis of SQSTM1 Disease Mutations
Several missense mutations in the SQSTM1 gene have been found in sporadic ALS patients. Particularly, the L341V missense disease mutation with a mutation in the LIR motif of SQSTM1 has been shown to decrease co-localization with the autophagy-related factor LC3 in cells. Various mutations in SQSTM1 are considered to be involved in abnormalities in cellular functions, including autophagy.
Future Challenges for SQSTM1
Knowledge about the molecular signaling pathways of SQSTM1 in the nervous system is still limited, and the relationship between SQSTM1 aggregate accumulation and disease onset remains mostly unknown. Future plans include further analysis focusing on the domain structure, molecular functional analysis, and the relationship between these domains and the development of neurodegenerative diseases caused by SQSTM1 mutations, including liquid-liquid phase separation (LLPS) and its association with SQSTM1 and autophagy.
Construction of a Novel Human Motor Neuron Model Using iPSCs
Induction of Motor Neuron Differentiation from ALS Patient iPSCs
Through collaborative research with Professor Hedeyuki Okano of Keio University, Professor Hiroaki Miyajima of Hamamatsu Medical University, and Professor Masashi Aoki of Tohoku University, our group has successfully established a culture method for inducing neurons with the characteristics of upper motor neurons from human iPSCs. Inducing differentiation of iPSCs into upper and lower motor neuron-like neurons has revealed selective cell death of upper motor neuron-like neurons in response to various stresses in type 2 ALS patients. These results suggest that human iPSC-derived upper motor neuron-like neurons could serve as a model for upper motor neuron degeneration in ALS patients. Currently, we are conducting cell function analysis focusing on ALS2 molecular network abnormalities to elucidate the molecular mechanisms of upper motor neuron degeneration in ALS patients.
Analysis of Axonal Organelle Dynamics Using Microfluidic Devices
To elucidate the association between ALS onset and transport abnormalities of autophagosomes and mitochondria, we are conducting an analysis of axonal organelle dynamics using microfluidic devices in collaboration with Professor Hiroshi Kimura of Micro/Nano Technology Center at Tokai University. Specifically, the microfluidic device controls the polarity of ALS mouse model and human iPSC-derived neuronal cells (refer to the figure below), allowing the temporal and quantitative measurement of transport of acidified vesicles and mitochondria, including autophagosomes, within axons. The goal of this research is to reveal the early abnormalities at the molecular and cellular levels before disease onset.
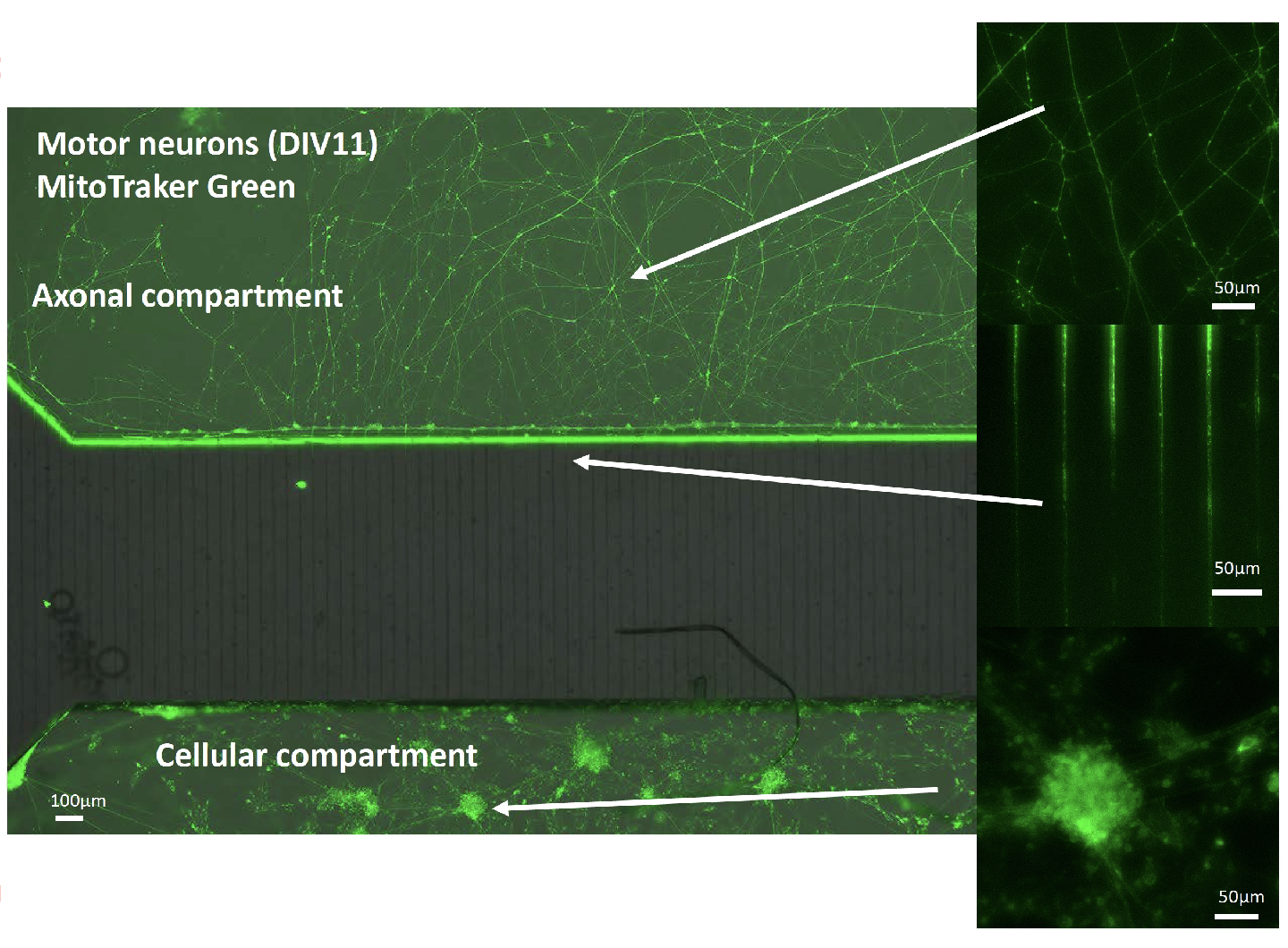
Other Research Topics
Development of a Novel ALS Therapeutic Drug Screening System
Focusing on the autophagy-lysosome system as a therapeutic target for ALS, we aim to establish a drug screening system based on the quantitative measurement of "degradation of abnormal proteins and maintenance of cellular organelle homeostasis through membrane vesicle transport." Currently, we are developing a monitoring method for autophagy-lysosome system at the molecular and cellular levels using mKeima-Red-LC3B transgenic mice.