研究内容
ALS2型原因遺伝子産物"ALS2"の分子機能解析
<ALS2 遺伝子とは?>
ALS2 遺伝子は、第2染色体長腕q33領域に位置する34個のエクソンからなる全長およそ80kbの遺伝子であり、常染色体劣性遺伝形式を示す3種類の運動ニューロン疾患(2型筋萎縮性側索硬化症;ALS2、若年発症型原発性側索硬化症;PLSJ、及び乳児期発症上行性遺伝性痙性対麻痺;IAHSP)の原因遺伝子である。ALS2 遺伝子は、6.5kbと2.6kbの2種類の転写産物を有し、特に神経系、筋肉、および腎臓組織で強く発現している。ALS2 転写配列内には、推定1657アミノ酸残基(184 kDa)からなるタンパク質をコードする翻訳領域が含まれている。<ALS2タンパク質とは?>
ALS2タンパク質(以下、ALS2と略、”Alsin”とも呼ばれる)には、3つの独立したグアニンヌクレオチド交換因子(guanine nucleotide exchanging factor; GEF)様ドメイン(RLD,DH/PH,およびVPS9)が存在する(下図参照)。N末端に存在するRLD(regulator of chromosome condensation 1–like domain)は、7つのブレードからなるβプロペラ構造を有する。RLDブレード5は、天然変性領域(intrinsically disordered region; IDR)の挿入により中断されている。DH/PH(Dbl-homology and pleckstrin-homology)にはRhoGEF活性はなく、またDH/PHの下流には細胞膜への結合に関与する8つのMORN(membrane occupation and recognition nexus)モチーフがある。C末端には、Rab5の活性化を触媒するVPS9(vacuolar protein sorting 9)ドメインが存在する。
<ALS2の機能>
これまでの研究により、ALS2タンパク質の構造と機能について、以下の点が明らかにされている。- ALS2は低分子量Gタンパク質Rab5の活性化因子である。
- ALS2は神経細胞を含めた培養細胞系においてはRab5の活性化を介してエンドソーム動態調節に重要な役割を果たしている。
- ALS2は神経細胞の軸索伸長に係わる調節因子である。
- Rac1活性化により、ALS2はマクロピノサイトーシスを介してエンドソームに局在する。
- ALS2はオートファジーによるタンパク質分解を調節する。
- ALS2はホモ4量体を形成し、その正常な高次構造がALS2のエンドソーム局在化及びRab5GEF活性に必須である。
- ALS2機能喪失は変異SOD1発現ALSマウスモデルの疾患症状を悪化させる。
- ALS2は低分子量Gタンパク質Rab17と結合し、Rab17陽性エンドソームの成熟とリサイクリングを制御する。
- ALS2のN末端RLD内に挿入されている天然変性領域(IDR)はALS2の細胞内局在及び複合体形成を制御している。
- ALS2は、中枢神経系において特異的な高分子ホモ複合体を形成している。
<ALS2に関する今後の課題>
現在、ALS2に結合する因子が多数同定されている。今後、ALS2の構造とその活性化の分子メカニズムを解析するとともに、新規の結合因子との機能的連関、ならびにエンドソーム-リソソーム系調節機能との関連を明らかにすることで、ALS2機能喪失により引き起こされる運動ニューロン疾患の発症メカニズムの解明を目指している。ALS関連因子SQSTM1/p62及び疾患変異体の分子機能解析
<SQSTM1/p62とは?>
SQSTM1 (sequestosome-1 あるいはp62とも呼ばれる)は、オートファジー経路におけるアダプタータンパク質としてタンパク質やオルガネラの分解経路に関わるとともに、Keap1と相互作用することでNrf2を介した酸化ストレス応答を促進する機能を持つ。さらに、SQSTM1はユビキチン化caspase-8の凝集によるアポトーシスの誘導経路にも関わることが知られている(下図参照)。また、SQSTM1は、大多数の孤発性ALS患者の脊髄において、凝集体の構成成分の一つとしてユビキチン化タンパク質とともに蓄積していることが明らかにされている。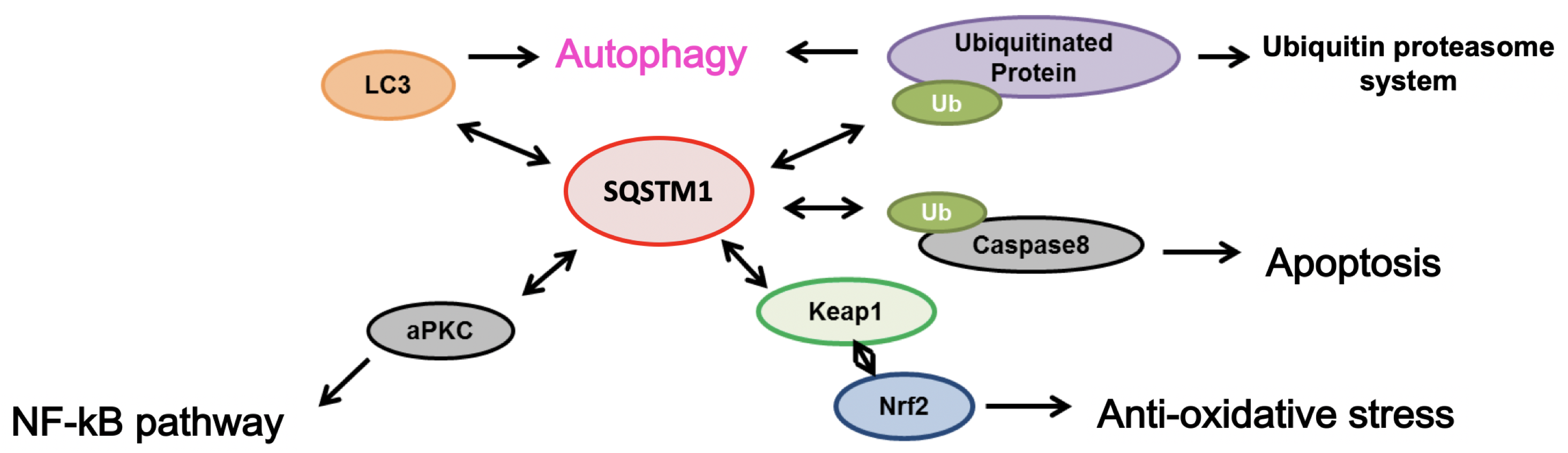
<SQSTM1 遺伝子変異とALS>
近年、家族性のみならず孤発性のALS及び前頭側頭型認知症(Frontotemporal Dementia; FTD)患者において、数多くのSQSTM1 遺伝子変異が同定されている。我々は、中国・四川大学および北京大学との共同研究により、孤発性ALS患者における複数の新たなSQSTM1 変異を同定している。<変異SOD1発現ALSマウスモデルにおけるSQSTM1/p62の役割>
これまでの研究により、SQSTM1の機能喪失によって変異SOD1発現型のALSマウスモデル(SOD1H46R)の疾患症状が顕著に悪化すること、ならびに全身的にSQSTM1を過剰発現させることによりSOD1H46Rの体重減少が加速化して疾患発症が早期化することが明らかにされている。また、一連の解析により、SQSTM1は疾患発症や進行に対して神経細胞における凝集体形成を促進して神経保護的に作用する反面、過剰に存在すると何らかの神経毒性を誘起することが示唆されている。これらのことから、ALS患者で同定されたSQSTM1 変異は、SQSTM1自体の神経細胞保護効果の減弱や毒性形質の獲得につながるものであると推定される。さらに、SQSTM1の機能喪失がALSマウスモデルの発症を早期化させるのみでなく、その早期化がALS2とSQSTM1両者の欠損により、相加的に加速することも明らかとなっている。
<SQSTM1疾患変異体の機能解析>
これまでの研究により、孤発性ALS患者からSQSTM1遺伝子における多くのミスセンス変異が見出されている。中でも、SQSTM1のLIRモチーフに変異を有するL341Vミスセンス疾患変異は、オートファジー関連因子であるLC3との細胞内での共存を低下させることが明らかにされている。SQSTM1の様々な変異がオートファジーを含めた細胞内機能の異常に関与している可能性があると考えられる。<SQSTM1に関する今後の課題>
神経系におけるSQSTM1分子シグナル系に関する知見は未だ限定的であり、SQSTM1凝集体蓄積と疾患発症との関連についてはほとんど不明である。SQSTM1を介したオートファジーによる選択的タンパク質・オルガネラ分解には、SQSTM1のPB1ドメインによる自己多量体化、LIRモチーフによるLC3との相互作用、およびUBAドメインを介したユビキチン化タンパク質との結合が重要であるとされる。今後、液-液相分離(LLPS)とSQSTM1およびオートファジーとの関連を含め、これらのドメイン構造に焦点を当てた分子機能解析、およびその変異による機能喪失機能喪失と神経変性疾患発症との関連についてさらに解析する計画である。iPS細胞を用いた新たなヒト運動ニューロンモデルの構築
<ALS患者iPS細胞からの運動ニューロンの分化誘導>
我々のグループは、慶應義塾大学・岡野栄之教授、浜松医科大学・宮嶋裕明教授、東北大学・青木正志教授との共同研究により、ヒトiPS細胞から世界初となる上位運動ニューロンの特徴を有する神経細胞の分化培養法の確立に成功している。また、iPS細胞から上位及び下位運動ニューロン様神経細胞に分化誘導させた結果、種々のストレスに対して2型ALS患者では上位運動ニューロン様神経細胞が選択的に細胞死に至ることも明らかにしている。これらの結果は、ヒトiPS細胞由来上位運動ニューロン様神経細胞がALS患者における上位運動ニューロン変性モデルになり得ることを示している。現在、ALS2分子ネットワーク異常を中軸にした細胞機能解析を行うことにより、ALS患者における上位運動ニューロン変性分子メカニズムの解明を目指している。<マイクロ流体デバイスを用いた軸索オルガネラ動態の解析>
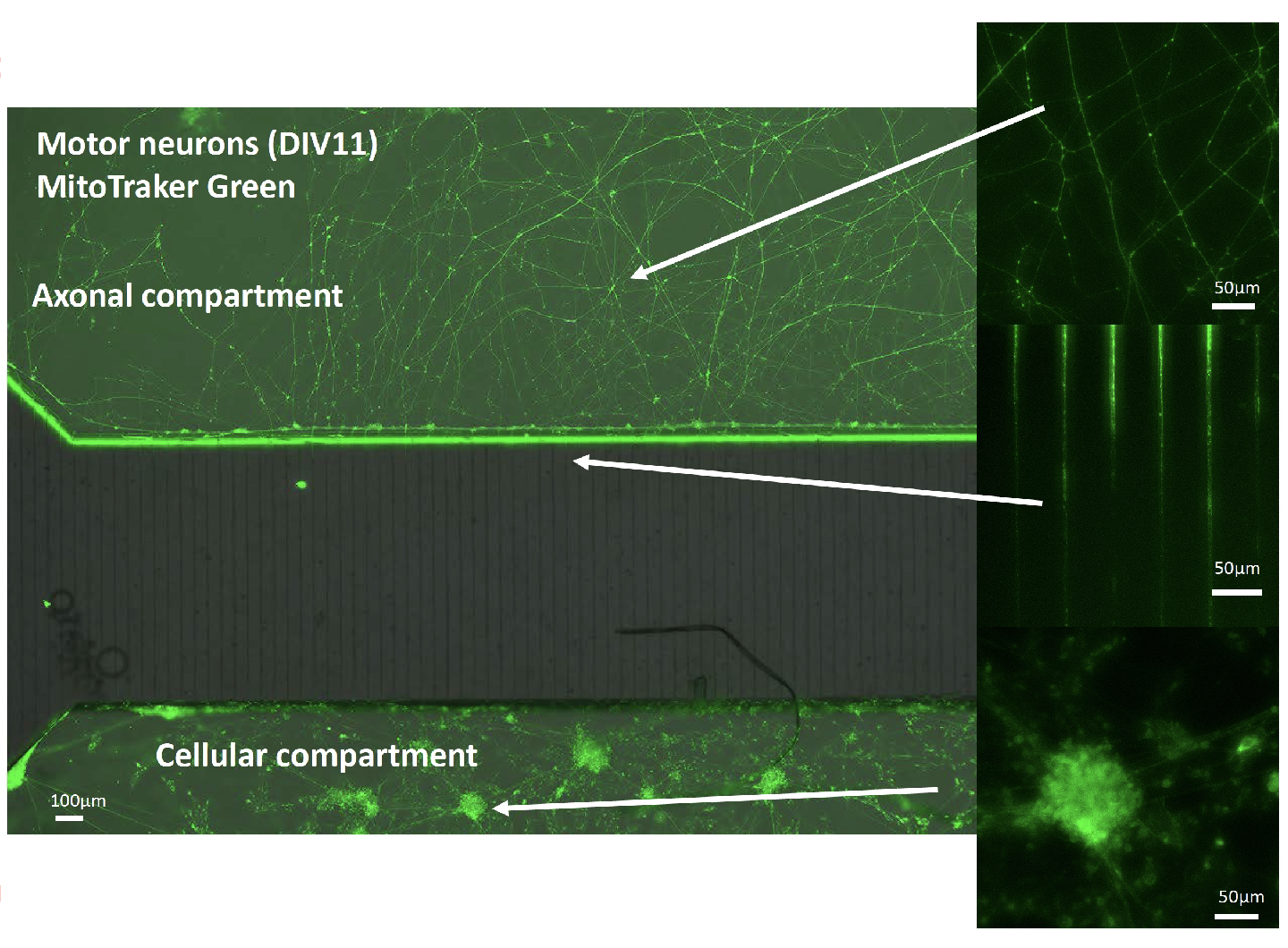
ALS疾患発症とオートファゴソームやミトコンドリアの輸送異常との関連を明らかにするため、東海大学マイクロナノ研究開発センター・木村啓志教授との共同研究により、マイクロ流体デバイスを用いた軸索オルガネラ動態の解析を行っている。具体的には、マイクロ流体デバイスによりALSマウスモデルおよびヒトiPS細胞由来の神経細胞の極性を制御し(下図参照)、軸索内のオートファゴソームを含む酸性小胞およびミトコンドリア輸送を経時的・定量的に測定する。この研究により、疾患発症前の分子・細胞レベルでの早期異常の実態を明らかにすることを目指している。